

2004年, Hayashi小组[22]首次将手性双烯作为配体用于铑催化的芳基硼酐对N-Ts保护的苯甲醛亚胺的不对称加成反应中, 利用C2对称双环[2.2.2]-辛二烯L7a, 反应可以取得非常出色的结果(最高98%收率, 99%ee), 而且底物适应性也很好, 亚胺以及硼酐芳环上取代基的空间和电子效应对反应的产率及选择性都没有明显影响(Eq. 17).与其它类型的配体相比, 手性双烯在这类反应中表现出更加优秀的催化活性和对映选择性.由于氮上Ts保护基通常难以脱除, 他们又发展了另外一类双环[3.3.1]-壬二烯骨架的手性双烯配体L8, 实现了对N-Ns (Ns=4-硝基苯磺酰基)亚胺的不对称芳基化, 取得了最高96%收率, 99% ee [23](Eq. 18).
对于反应产物的立体化学, 如Eq. 19所示, 推测转金属化后得到的芳基铑物种与亚胺底物可能有两种配位方式, 由于过渡态TS-1中亚胺氮上的磺酰基与配体双键上的苯基有较大的空间位阻, 所以TS-2为优势过渡态, 芳基迁移后得到主要为S构型的产物[22].
基于双环[2.2.2]-己二烯骨架, Hayashi小组[24]之后又成功发展了一类C1对称的手性双烯配体L9a.该配体在芳基硼酐对芳基醛亚胺的不对称加成反应中同样显示出了非常好的催化活性, 催化剂用量甚至可以低至0.3 mol%, 反应收率和对映选择性均极其优异(90%~98%收率, 97%~99.5% ee) (Eq. 20).该配体具有高催化活性的原因可能在于引入吸电子的酯基后, 双键更加缺电子, 有利于转金属化, 由于转金属化是反应的决速步, 所以该配体表现出更好的催化活性.
利用双烯配体L9b, 在相同的反应体系下, Woodward等[25]实现了磺酰二亚胺底物的双不对称芳基化, 以较高的立体选择性得到相应的手性磺酰二胺化合物.这些产物在吡啶/水体系回流条件下可以脱去二氧化硫得到手性二芳基甲胺, 但反应有不同程度的消旋化发生(Eq. 21).
为避免上述保护基脱除过程中产物的部分消旋化, 该小组[26]又设计了在亚胺氮上引入另外一种能在温和条件下方便地脱除的N-叔丁基磺酰胺基(Eq. 22).但在配体L9b促进的芳基化反应条件下, 部分底物由于亚胺水解导致收率大大降低, 有意思的是, 当换用C2对称的手性双烯L7a为配体时, 反应可以取得较高的收率.
基于C1对称的手性双烯配体L9b表现出来的高催化活性, Morimoto等[27]设计了一个串联的不对称芳基化-氨羰基化环合反应用于手性异吲哚啉类化合物的合成.该方法避免了一氧化碳气体的使用, 利用芳基醛或者多聚甲醛为羰基源, 反应中间体无需分离, 底物适应性好, 产物的收率和光学纯度均不错(Eq. 23).
2007年, Lin和Xu等[28]报道了一类结构简单的基于双环[3.3.0]结构的全新手性双烯配体L10, 分子中两个顺并的环戊烯环在空间上呈楔形, 在催化过程中, 两个烯烃双键与金属铑配位可以提供一个对反应的选择性控制非常有利的手性环境, 尤其在铑催化的芳香醛亚胺的不对称芳基加成中能取得极其优秀的对映选择性, 几乎所有产物的光学纯度都在98%~99% ee (Eq. 24).
带邻位酯基的苯甲醛亚胺同样能很高效地发生反应, 在相同的反应条件下经不对称芳基加成后原位内酰氨化, 可以直接合成高光学纯度的手性异吲哚啉化合物(Eq. 25), 而这类化合物中3-位手性的构建用其它催化方法通常是比较困难的.
进一步的研究发现, 利用硼酸/KHF2/H2O的组合反应体系[29], 在手性双环[3.3.0]-辛二烯配体L10促进下, 可以实现对N-Ns亚胺的高对映选择性芳基加成, 以最高96%收率和99% ee得到相应的二芳基甲胺产物[30] (Eq. 26).
随后, 该催化体系又被成功用于芳基硼酸对吲哚醛亚胺的不对称加成反应中, 产物的光学纯度最高可以达到99% ee[31] (Eqs. 27, 28).值得一提的是, 反应能很好地适用于各种缺电性底物, 为合成传统的Friedel-Crafts策略难以实现的α-芳基3-吲哚甲胺类化合物提供了一个新的方法.同时, 对于更加挑战的2-吲哚醛亚胺, 该反应体系也能高效地获得目标芳基化产物(72%收率, 98.5% ee).
2010年, Lin, Feng等[32]报道了另外一类C1对称的同时具有桥环和并环结构的手性双烯配体, 发现这类配体也能高效催化芳基硼酸对醛亚胺的不对称加成, 反应几乎都能取得定量的收率, 但立体选择性较之前C2对称的手性双烯配体均有明显的下降(Eq. 29).
脂肪亚胺的不对称芳基加成报道很少, 一般来说, 需要找到一个活性很高的催化剂能促使芳基化反应顺利发生, 同时反应体系中要避免使用酸或碱以减少副反应的发生. 2011年, Lin小组[33]发现利用铑/烯烃配合物[Rh(L10)(OH)]2可以同时满足如上两个条件, 体系中分子筛的加入也能进一步提高收率.反应的底物适应性较好, 并且收率和对映选择性都很出色(87%~99%收率, 91%~99% ee).结合反应后分子内取代环化策略, 该方法可以用于手性2-芳基吡咯烷及2-芳基哌啶类化合物的高效不对称合成(Eqs. 30, 31).此外, 该催化体系同样适用于N-Ns亚胺, 反应都可以取得良好的收率(87%~99%)和极其优秀的对映选择性(>99% ee).
2010年, Du小组[34]报道了一类基于手性联萘骨架的开链的手性双烯配体在铑催化的芳基醛亚胺的不对称芳基加成反应中的应用.研究发现, 配体分子中联萘骨架上3, 3’位的取代对催化剂的活性和立体选择性有明显的影响, 当3, 3’位无取代时, 该类双烯配体几乎不能催化反应.从反应的底物适应性来看, 该催化体系似乎仅适用于给电性很强的对位烷氧基取代的苯硼酸; 当选用对甲基苯硼酸时, 收率下降至48% (Eq. 32).
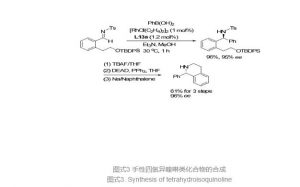
2014年, Wu小组[35]报道了另一类C1对称手性双烯配体L13a促进的铑催化的芳基硼酸对芳香醛亚胺的不对称加成反应, 该反应同时适用于N-Ts及N-Ns亚胺并且底物适应性较好; 催化剂具有较高的反应活性, 反应1 h便可完全, 可以以56%~99%收率, 90%~99% ee得到手性二芳基甲胺产物.在亚胺底物中引入合适的官能团, 其加成产物经过几步简单转化可用于手性四氢异喹啉类化合物的合成(Eq. 33, Scheme 3).
化学慧定制合成摘录




