

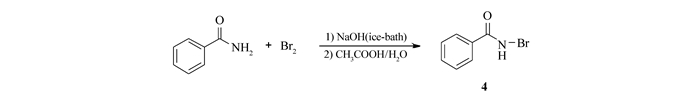 Scheme 2. Preparation of N-bromobenzamide
Scheme 2. Preparation of N-bromobenzamide 2-噁唑啉是一类重要的杂环化合物,通过其区域选择性和立体选择性开环反应可形成多种功能化衍生物,因而在有机合成中是一类非常重要的合成中间体[1–2]。事实上很多人工合成的化合物和天然产物中都含有2-噁唑啉结构单元,并且由于此种结构单元的存在而表现出强烈的生物活性[3–4]。此外作为配体,手性2-噁唑啉衍生物已被广泛地应用于催化不对称合成中[5–6]。由于2-噁唑啉衍生物已在诸多领域表现出重要的应用价值,它的合成长期为有机化学家和生物化学家所关注[1–7]。在过去的几十年里,已有很多文献报道了合成2-噁唑啉衍生物的方法,这些方法大部分由邻位氨基醇分别与羧酸[8]、酯[9–10]、腈[11–12]、醛[13–14]以及Vilsmeier试剂[15]缩合,先得到β-羟基酰胺,再由β-羟基酰胺进行分子内脱水得到相应的2-噁唑啉衍生物。然而,以上方法不同程度存在着不足,有的反应条件苛刻,有的反应时间太长,有的反应对环境污染较大等。因此,研究由易得的反应底物出发,在温和反应条件下,高效合成具有多功能基团2-噁唑啉衍生物新方法显得尤为必要。
虽然2-噁唑啉衍生物可以由烯烃直接合成,但能用于合成的烯烃范围往往受到限制。事实上,仅有由简单烯烃合成2-噁唑啉衍生物的文献报道[16–17]。本课题组在研究缺电子烯烃的氨卤加成反应中[18–23]发现,α-氰基肉桂酸乙酯可与N-溴代苯甲酰胺(N-溴代芳香酰胺)发生反应并形成相应2-噁唑啉衍生物。经过系统研究, 建立了在弱碱促进下, 在丙酮溶剂中, 由α-氰基肉桂酸乙酯衍生物和N-溴代苯甲酰胺合成相应2-噁唑啉衍生物的新方法。
1 实验部分
1.1 仪器和试剂
XT5A型显微熔点测定仪(微电脑控制型, 熔点未经校正, 北京市科仪电光仪器厂); BS224S型电子天平(德国赛多利斯集团); AVANCE 400 MHz型超导傅里叶数字化核磁共振仪(NMR, 瑞士Bruker公司); GC-MSQPZ2010型质谱仪(MS, 日本岛津公司)。
芳醛、氰基乙酸乙酯、苯甲酰胺均购自国药集团化学试剂有限公司;所用其它试剂均为分析纯, 使用前未经纯化处理。
1.2 实验方法
1.2.2 N-溴代苯甲酰胺(4)的合成
根据文献[34]合成氮溴代苯甲酰胺(Scheme 2)。向一个盛有150 mL水的锥形瓶中,加入固体氢氧化钠9.2 g(0.23 mol),待固体完全溶解并冷却后,冰浴下,边搅拌,边滴加2.3 mL(14.4 g, 0.09 mol)的液溴,得到浅黄色的NaBrO溶液备用。
Scheme 2. Preparation of N-bromobenzamide
将10 g苯甲酰胺研细,分3~4次加入到上述配好的NaBrO溶液中(操作在冰浴、搅拌下进行)。待苯甲酰胺全部加完后,再搅拌约10 min。此时溶液的黄色褪去,然后将溶液快速过滤到加有9 mL冰乙酸的25 mL冰水中,析出浅黄色固体, 过滤,并用水洗涤固体2~3次。将得到的固体先用40~50 mL的氯仿加热到沸腾使之溶解,然后加入等体积的石油醚,溶液变浑浊。放置冰箱中过夜,析出白色固体,过滤,真空干燥得到N-溴代苯甲酰胺(4) 4.2 g(收率51%),mp 129~131 ℃(lit.[34]129~131 ℃)。1H NMR(400 Hz, CDCl3), δ:6.79(s, 1H, NH), 7.39~7.56(m, 3H, ArH), 7.77~7.81(m, 2H, ArH)。
1.2.4 中间体13的结构表征
淡黄色固体,mp 213~215 ℃。1H NMR(400 MHz, CDCl3), δ:7.84(s, 2H, ArH), 7.56(d, J=8.0 Hz, 1H, NH), 7.49(t, J=8.0 Hz, 2H, ArH), 7.43(d, J=8.0 Hz, 2H, ArH), 7.21(d, J=8.0 Hz, 2H, ArH), 6.93(d, J=12 Hz, 1H, ArH), 6.15(d, J=12 Hz, 1H, CH), 4.23(, J=8.0 Hz, 2H, OCH2), 2.36(s, 3H, ArCH3), 1.22(t, J=8.0Hz, 3H, CH2CH3); 13C NMR(100 MHz, CDCl3), δ:166.8, 162.4, 139.8, 133.5, 132.2, 131.1, 129.7, 128.8, 127.8, 127.2, 114.7, 64.67, 57.66, 49.19, 21.13, 13.53;HRMS(ESI)计算值C20H19BrN2NaO3[M+Na]+:437.0477, 实测值:437.0471。
以上化合物的波谱图和高分辩率质谱图见辅助材料。
1.2.3 2-噁唑啉衍生物的合成
在一个25 mL干燥的圆底烧瓶中,依次加入α-氰基肉桂酸乙酯衍生物(3) 2.0 mmol,N-溴代苯甲酰胺(4) 4.0 mmol, Na2CO3 2.2 mmol,丙酮10.0 mL, 室温搅拌,薄层色谱(TLC)跟踪反应,直到化合物3完全消失或产物不再增加为止。反应完全后,向反应液中加入20 mL乙酸乙酯,用饱和食盐水洗涤3次(每次10 mL),再用水洗涤3次(每次10 mL), 有机相用无水硫酸钠干燥, 减压蒸掉有机溶剂, 混合物用柱色谱分离(V(石油醚):V(乙酸乙酯)=12:1),得到产物5。合成路线见Scheme 3。
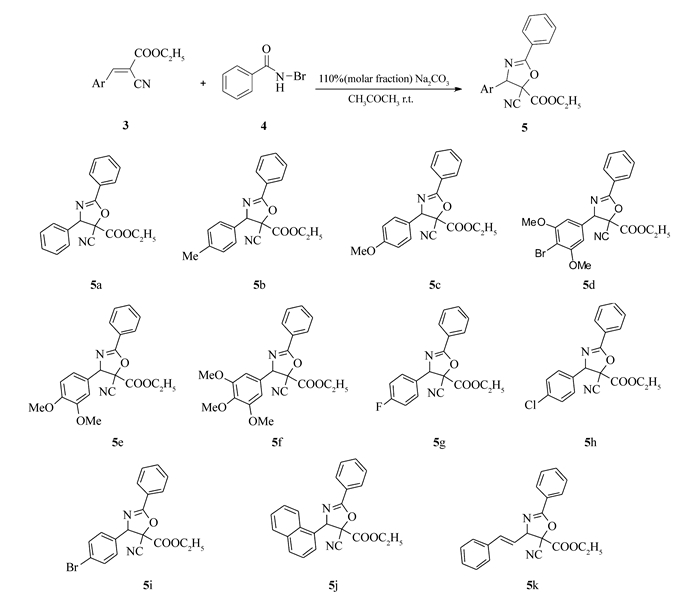 Scheme 3. Synthesis and structures of 2-oxazoline derivatives(5a~5k)
Scheme 3. Synthesis and structures of 2-oxazoline derivatives(5a~5k)
产物5a:黄色固体,收率72%,mp 60~61 ℃。1H NMR(400 MHz, CDCl3), δ:8.06(d, J=8.0 Hz, 2H, ArH), 7.40(t, J=8.0 Hz, 1H), 7.37(d, J=4.0Hz, 4H, ArH), 7.34(t, J=4.0 Hz, 3H, ArH), 5.76(s, 1H, CH), 4.32(q, J=4.0Hz, 2H, OCH2), 1.28(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:165.0, 163.0, 136.0, 132.9, 129.5, 129.1, 128.9, 127.5, 125.5, 113.4, 82.41, 80.11, 64.45, 14.04。电喷雾离子化-高分辨质谱HRMS(ESI)计算值C19H16N2NaO3[M+Na]+:343.1059, 实测值:343.1051。
化合物5b:黄色固体,收率82%,mp 115~116 ℃。1H NMR(400 MHz, CDCl3), δ:8.08(d, J=4.0 Hz, 2H, ArH), 7.57(s, 1H, ArH), 7.47 (s, 2H, ArH), 7.23 (s, 4H, ArH), 5.72(s, 1H, CH), 4.45(q, J=4.0 Hz, 2H, OCH2), 2.37 (s, 3H, ArCH3), 1.40(t, J=4.0 Hz, 3H, CH3); 13C NMR(100 MHz, CDCl3), δ:165.1, 162.9, 139.4, 132.8, 132.7, 129.7, 129.0, 128.7, 127.2, 125.4, 113.3, 82.37, 79.96, 64.30, 21.32, 14.04;HRMS(ESI)计算值C20H18N2NaO3[M+Na]+:357.1215, 实测值:357.1207。
产物5c:白色固体,收率79%,mp 176~177 ℃。1H NMR(400 MHz, CDCl3), δ:8.08(d, J=4.0 Hz, 2H, ArH), 7.50(s, 1H, ArH), 7.47(s, 2H, ArH), 7.27(s, 2H, ArH), 6.95(d, J=8.0 Hz, 2H, ArH), 5.71(s, 1H, CH), 4.45(q, J=4.0 Hz, 2H, OCH2), 3.81(s, 3H, ArOCH3), 1.41(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:164.0, 161.7, 159.4, 131.7, 127.9, 127.7, 127.6, 126.8, 124.4, 113.3, 112.3 81.36, 78.69, 63.24, 54.25, 13.00;HRMS(ESI)计算值C20H18N2NaO4[M+Na]+:373.1159, 实测值:373.1157。
产物5d:白色固体,收率70%,mp 125~126 ℃。1H NMR(400 MHz, CDCl3), δ:8.00(d, J=8.0 Hz, 2H, ArH), 7.52(s, 1H, ArH), 7.43(t, J=12 Hz, 2H, ArH), 6.50(d, J=4.0 Hz, 1H, ArH), 6.46(d, J=4.0 Hz, 1H, ArH), 6.27(s, 1H, CH), 4.38(q, J=4.0Hz, 2H, OCH2), 3.82(s, 3H, ArOCH3), 3.70(s, 3H, ArOCH3), 1.34(t, J=4.0 Hz, 3H, CH3); 13C NMR(100 MHz, CDCl3), δ:164.9, 163.0, 160.2, 156.9, 137.7, 132.8, 129.0, 128.8, 125.3, 112.9, 105.8, 103.8, 100.4, 82.13, 79.28, 64.43, 56.43, 55.63, 13.94;HRMS(ESI)计算值C21H20BrN2O5[M+H]+:459.0556, 实测值:459.0554。
产物5e:浅黄色油状,收率87%。1H NMR(400 MHz, CDCl3), δ:7.98(d, J=8.0 Hz, 2H, ArH), 7.46(s, 1H, ArH), 7.37(s, 2H, ArH), 6.82(d, J=8.0Hz, 2H, ArH), 6.79(t, J=8.0Hz, 1H, ArH), 5.62(s, 1H, CH), 4.33(q, J=4.0 Hz, 2H, OCH2), 3.77(s, 6H, ArOCH3), 1.28(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:165.0, 162.7, 150.0, 149.3, 132.8, 129.0, 128.7, 128.1, 125.4, 120.1, 113.3, 111.3, 110.4, 82.39, 79.85, 64.31, 55.99, 55.87, 13.99;HRMS(ESI)计算值C21H21N2O5[M+H]+:381.1450, 实测值:381.1451。
产物5f:浅黄色固体,收率90%,mp 98~99 ℃。1H NMR(400 MHz, CDCl3), δ:8.09(d, J=4.0 Hz, 2H, ArH), 7.89(s, 1H, ArH), 7.51(t, J=4.0 Hz, 2H, ArH), 6.57(s, 2H, ArH), 5.69(s, 1H, CH), 4.48(q, J=4.0 Hz, 2H, OCH2), 3.87(s, 9H, ArOCH3), 1.43(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:165.0, 162.9, 153.6, 139.0, 132.9, 131.0, 129.0, 128.8, 125.3, 113.2, 104.7, 82.31, 80.02, 64.38, 60.91, 56.27, 14.05;HRMS(ESI)计算值C22H21N2O6[M+H]+:411.1556, 实测值:411.1558。
产物5g:浅黄色油状,收率64%。1H NMR(400 MHz, CDCl3), δ:7.95(d, J=4.0 Hz, 2H, ArH), 7.45(t, 1H, ArH), 7.23(d, J=4.0Hz, 2H, ArH), 7.01(d, J=4.0 Hz, 2H, ArH), 6.98(s, 2H, ArH), 5.64(s, 1H, CH), 4.30(dd, J=8.0 Hz, 4.0 Hz, 2H, OCH2), 1.25(t, J=4.0 Hz, 3H, CH3); 13C NMR(100 MHz, CDCl3), δ:164.8, 164.2, 163.1, 162.5, 132.9, 131.8, 131.8, 129.4, 129.3, 129.0, 128.8, 125.2, 116.1, 115.9, 113.2, 82.29, 79.29, 64.49, 14.00;HRMS(ESI)计算值C19H15FN2NaO3[M+Na]+:361.0969, 实测值:361.0956。
产物5h:浅黄色固体,收率67%,mp 94~95℃。1H NMR(400 MHz, CDCl3), δ:8.07(d, J=4.0 Hz, 2H, ArH), 7.58(s, 1H, ArH), 7.48(d, J=4.0 Hz, 2H, ArH), 7.43(d, J=8.0 Hz, 2H, ArH), 7.30(d, J=8.0 Hz, 2H, ArH), 5.74(s, 1H, CH), 4.44(q, J=4.0Hz, 2H, OCH2), 1.40(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:164.8, 163.3, 135.5, 134.4, 132.9, 129.2, 129.0, 128.8, 125.2, 113.1, 82.16, 79.28, 64.52, 14.03;HRMS(ESI)计算值C19H15ClN2NaO3[M+Na]+:377.0663, 实测值:377.0662。
产物5i:白色固体,收率70%,mp 107~109 ℃。1H NMR(400 MHz, CDCl3), δ:8.07(d, J=8.0 Hz, 2H, ArH), 7.56(s, 3H, ArH), 7.48(s, 2H, ArH), 7.24(t, J=4.0 Hz, 2H, ArH), 5.72(s, 1H, CH), 4.45(q, J=4.0 Hz, 2H, OCH2), 1.41(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:164.7, 163.3, 134.9 134.8, 132.9, 132.2, 129.1, 129.0, 128.8, 125.1, 123.7, 113.1, 82.07, 79.33, 64.53, 14.04;HRMS(ESI)计算值C19H15BrN2NaO3[M+Na]+:421.0158, 实测值:421.0160。
产物5j:浅黄色固体,收率65%,mp 145~146℃。1H NMR(400 MHz, CDCl3), δ:8.06(d, J=4.0 Hz, 2H, ArH), 7.85(q, J=4.0Hz, 3H, ArH), 7.50(s, 1H, ArH), 7.46(s, 2H, ArH), 7.42(s, 4H, ArH), 6.58(s, 1H, CH), 4.41(dd, J=4.0, 8.0 Hz, 2H, OCH2), 1.34(t, J=4.0 Hz, 3H, CH3);13C NMR(100 MHz, CDCl3), δ:165.3, 163.1, 133.9, 132.8, 131.6, 130.9, 130.0, 129.4, 129.0, 128.8, 126.8, 126.0, 125.9, 125.5, 125.4, 122.2, 113.0, 82.40, 75.88, 64.56, 13.98;HRM(ESI)计算值C23H18FN2NaO3[M+Na]+:393.1215, 实测值:393.1210。
产物5k:浅黄色油状,收率75%。1H NMR(400 MHz, CDCl3), δ:7.95(s, 2H, ArH), 7.49~7.46(m, 1H, ArH), 7.42~7.36(m, 4H, ArH), 7.28~7.23(m, 2H, ArH), 7.20(d, J=7.3 Hz, 1H, ArH), 6.76(d, J=15.6 Hz, 1H, CH═), 6.28(q, J=4.0 Hz, 1H, CH═), 5.20(d, J=4.0 Hz, 1H, CH), 4.33(q, J=4.0Hz, 2H, OCH2), 1.29(t, J=4.0 Hz, 3H); 13C NMR(100 MHz, CDCl3), δ:164.7, 162.4, 136.1, 135.7, 132.8, 128.9, 128.7, 128.7, 128.7, 127.1, 125.4, 123.3, 113.4, 81.02, 77.33, 64.33, 14.02;HRMS(ESI)计算值C21H19N2O3[M+H]+:347.1396, 实测值:347.1396。
产物10:白色固体,mp 147~149 ℃。1H NMR(400 MHz, CDCl3), δ:8.36(d, J=12 Hz, 2H, ArH), 8.29(d, J=12 Hz, 2H, ArH), 7.48(d, J=8.0 Hz, 3H, ArH), 7.36(d, J=8.0 Hz, 2H, ArH), 5.84(s, 1H, CH), 4.51(t, J=8.0Hz, 2H, OCH2), 1.46(t, J=8.0 Hz, 3H, CH3); 13C NMR(100 MHz, CDCl3), δ:164.5, 161.1, 150.3, 135.0, 130.9, 130.1, 129.7, 129.3, 129.1, 127.2, 123.9, 112.6, 82.54, 80.05, 64.65, 13.97;HRMS(ESI)计算值C19H15N3NaO5[M+Na]+:388.0909, 实测值:388.0918。
产物11:白色固体,mp 50~52 ℃。1H NMR(300 MHz, CDCl3), δ:7.44~7.28(m, 5H, ArH), 5.53(s, 1H, CH), 4.45(q, 2H, OCH2), 2.25(s, 3H, CH3), 1.41(t, J=6.0Hz, 3H, CH2CH3); 13C NMR(75.5 MHz, CDCl3), δ:165.0, 164.1, 135.5, 129.4, 128.9(2), 127.1(2), 113.0, 82.1, 79.7, 64.3, 14.0, 13.6;HRMS(ESI)计算值C14H14NaN2O3[M+Na]+:281.0897, 实测值:281.0892。
产物12:淡黄色油状。1H NMR(400 MHz, CDCl3), δ:8.31(d, J=8.0 Hz, 2H, ArH), 7.95(d, J=8.0 Hz, 2H, ArH), 7.49(s, 2H, ArH), 7.35(t, J=4.0 Hz, 3H, ArH), 6.11(d, J=9.5 Hz, 1H, CH), 4.29(q, J=8.0 Hz, 4H, OCH2), 1.26(t, J=4.0 Hz, 6H, CH2CH3); 13C NMR(100 MHz, CDCl3), δ:171.1, 166.4, 163.8, 149.8, 139.5, 135.80, 128.9, 128.4, 128.3, 123.9, 63.94, 60.37, 21.01, 14.17;HRMS(ESI)计算值C21H21N2O7[M+H]+413.1349, 实测值:413.1362。
1.2.1 α-氰基肉桂酸乙酯衍生物(3)的合成
α-氰基肉桂酸乙酯衍生物依照参考文献[24]合成,合成路线见Scheme 1。
 Scheme 1. Synthesis of α-cyanocinnamate derivatives
Scheme 1. Synthesis of α-cyanocinnamate derivatives
化合物3a:白色固体,mp 49~50 ℃(lit.[25]49~50 ℃); 1H NMR(400 Hz, CDCl3), δ:8.26(s, 1H, CH═), 7.99(d, J=6.9 Hz, 2H, ArH), 7.48~7.59(m, 3H, ArH), 4.40(q, J=7.2 Hz, 2H, OCH2), 1.41(t, J=7.2Hz, 3H, CH3)。
化合物3b:黄色固体,mp 92.5~93.5 ℃(lit.[26]93.6 ℃); 1H NMR(400 Hz, CDCl3), δ:8.21(s, 1H, CH═), 7.90(d, 2H, J=7.5 Hz, ArH), 7.30(d, 2H, J=7.5 Hz, ArH), 4.37(q, 2H, J=6.0 Hz, CH2), 2.43 (s, 3H, ArCH3), 1.39(t, 3H, J=6.8 Hz, CH3)。
化合物3c:浅黄色固体,mp 80~82 ℃(lit.[27]82~84 ℃); 1H NMR(400 MHz, CDCl3), δ:8.19(s, 1H, CH═), 8.02(d, J=8.9 Hz, 2H, Ar), 7.01(d, J=8.9 Hz, 2H, Ar), 4.38(q, J=7.1 Hz, 2H, CH2), 3.91(s, 3H, CH3O), 1.40(t, J=7.1 Hz, 3H, CH3)
化合物3d:黄色固体,mp 85~86 ℃(lit.[28]85~87 ℃); 1H NMR(400 MHz, CDCl3), δ:8.17(s, 1H, CH═), 8.08 (s, 2H, ArH), 7.98(s, 1H, ArH), 4.38(q, J=7.2 Hz, 2H, CH2), 3.92 (s, 6H, CH3O), 1.39(t, J=7.1 Hz, 3H, CH3)。
化合物3e:黄色固体,mp 154~155 ℃(lit.[29]154~156 ℃); 1H NMR(400 MHz, CDCl3), δ:8.15(s, 1H, CH═), 8.00(s, 2H, ArH), 7.01(s, 1H, ArH), 4.38(q, J=7.1 Hz, 2H, CH2), 3.92(s, 3H, CH3O), 3.91(s, 3H, CH3O), 1.39(t, J=7.1 Hz, 3H, CH3)。
化合物3f:黄色固体,mp 81~82 ℃(lit.[30]81.5~82.5 ℃); 1H NMR(400 MHz, CDCl3), δ:8.13(s, 1H, CH═), 8.00(s, 2H, ArH), 4.38(q, J=7.2 Hz, 2H, CH2), 3.92(s, 9H, CH3O), 1.39(t, J=7.1 Hz, 3H, CH3)。
化合物3g:白色固体,mp 95~96 ℃(lit.[31]94~96 ℃); 1H NMR(400 MHz, CDCl3), δ:8.25(s, 1H, CH═), 8.00(d, J=8.6 Hz, 2H, ArH), 7.56(d, J=8.3 Hz, 2H, ArH), 4.35(q, J=7.0 Hz, 2H, OCH2), 1.39(t, J=6.9 Hz, 3H, CH3)。
化合物3h:白色固体,mp 94~96 ℃(lit.[24]93 ℃); 1H NMR(400 Hz, CDCl3), δ:8.20(s, 1H, CH═), 7.95(d, J=8.7 Hz, 2H, ArH), 7.50(d, J=8.7 Hz, 2H, ArH), 4.40(q, J=6.9 Hz, 2H, OCH2), 1.40(t, J=6.9 Hz, 3H, CH3)。
化合物3i:白色固体,mp 97~98 ℃(lit.[32]97.5 ℃); 1H NMR(400 Hz, CDCl3), δ:8.18(s, 1H, CH═), 7.93(d, J=8.7 Hz, 2H, ArH), 7.48(d, J=8.7 Hz, 2H, ArH), 4.42(q, J=6.9 Hz, 2H, OCH2), 1.38(t, J=6.7Hz, 3H, CH3)。
化合物3j:黄色固体,mp 81~83 ℃(lit.[33]81~82 ℃); 1H NMR(400 Hz, CDCl3), δ:8.13(s, 1H, CH═), 7.80~7.68(m, 4H, ArH), 7.64~7.44(m, 3H, ArH), 4.32(q, J=7.0 Hz, 2H, OCH2), 1.40(t, J=7.5Hz, 3H, CH3)。
化合物3k:黄色固体,mp 110.4~112 ℃(lit.[24]111~112 ℃); 1H NMR(CDCl3), δ:8.00(dd, J=3.9, 6.9 Hz, 1H, CH═), 7.57~7.61(m, 2H, Ar), 7.41~7.44(m, 3H, Ar), 7.26(q, J=3.6 Hz, 2H, CH═), 4.35(q, J=7.2 Hz, 2H, OCH2), 1.38(t, J=7.2Hz, 3H, CH3)。
2 结果与讨论
2.1 反应物料比的确定
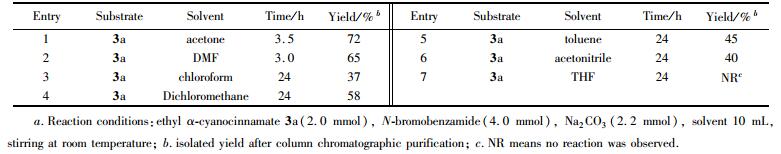
为了优化反应条件,首先用α-氰基肉桂酸乙酯(3a)为底物,以N-溴代苯甲酰胺(4)为反应试剂, 在Na2CO3(110%摩尔分数)促进下,以二氯甲烷作为溶剂,对反应的物料比进行了考察。发现当底物与反应试剂的摩尔比为n(3a):n(4)=1:1时,反应24 h得到了32%的收率。当n(3a):n(4)=1:1.5时, 收率提高到了45%;当n(3a):n(4)=1:2时,收率可提高到58%,继续提高反应试剂4的量,反应收率没有明显提高,仅仅是反应时间缩短了。因此,最后确定该反应的最佳物料比为n(3a):n(4)=1:2。
2.2 溶剂对反应的影响
在确定了物料比之后,仍然用α-氰基肉桂酸乙酯(3a)为底物,以N-溴代苯甲酰胺为反应试剂, 在Na2CO3(相对于底物3a为110%摩尔分数)促进下,对常用溶剂进行了筛选,结果见表 1。由表 1可以看出,该反应在丙酮、甲苯、乙腈、二甲基甲酰胺(DMF)、二氯甲烷,氯仿中均能发生, 其中丙酮效果最好,反应只需要3.5 h就可以得到72%的收率。DMF虽然在3 h可以完成反应, 但收率不及丙酮。而四氢呋喃(THF)不是该反应的有效溶液剂。
 |
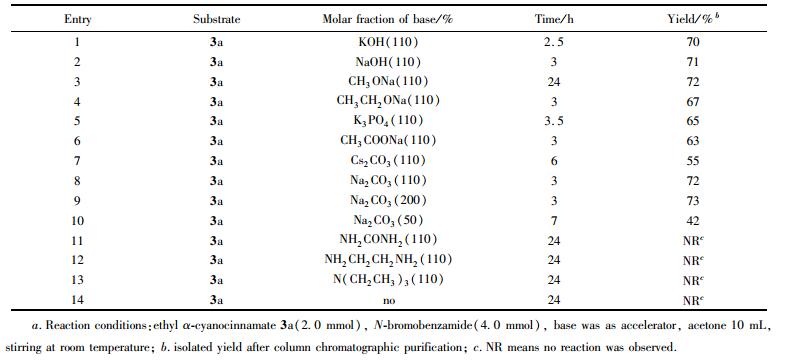
2.3 碱对反应的影响
当物料比为n(3a):n(4)=1:2,溶剂为丙酮时,用同样的模板反应对常用碱的种类和用量进行了筛选, 结果见表 2。可知, 各种无机碱均能有效促进该反应(表 2, Entries 1~8), 而且强碱和弱碱的效果差别并不明显, 然而有机碱并非该反应的有效促进剂(Entries 11~13)。综合考虑, 确定本反应的促进剂为Na2CO3。对Na2CO3的用量也进行了考察,结果表明, 110%摩尔分数Na2CO3为必要的用量(表 2, Entries 8~10)。对比实验表明, 在不加碱时该反应不能发生(Entry 14), 这说明碱起着重要的促进作用。
 |
2.4 底物结构对反应的影响
实验结果表明,当n(3a~3k):n(4):n(Na2CO3)=1:2:1.1时,丙酮作溶剂,几乎所有的α-氰基肉桂酸乙酯衍生物都能顺利地与N-溴代苯甲酰胺发生反应,得到相应的目标产物, 收率64%~90%, 见表 3。由表 3可知, 当底物苯环上没有任何取代基时(3a),反应收率为72%(表 3, Entry 1), 当与碳碳双键直接相连的苯环上连有吸电子基时(表 3, Entries 7~9),反应收率为64%~70%。当与碳碳双键直接相连的苯环上连有给电子基时, 收率比前者有所提高(表 3, 82%~90%, Entries 2~3, Entries 5~6)。芳基为1-萘基和苯乙烯基时, 也能得到较高的收率(表 3, entries 10~11)。以上实验结果表明, 底物苯环上取代基的性质对底物反应活性有重要影响, 这种电子效应的影响,表现为碳碳双键上电子密度越高,反应的活性越强。
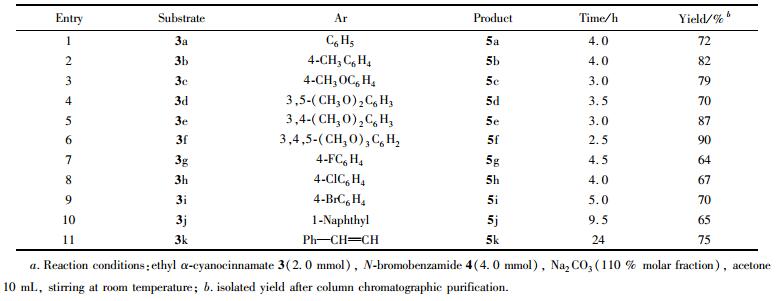 |
有趣的是,当Ar=3, 5-(CH3O)2C6H3时,得到的产物结构中除了在底物双键处形成正常的噁唑啉环外, 底物苯环的4位氢也被溴取代(5d), 见Scheme 4,说明在反应过程中一定存在溴正离子。根据底物和反应试剂的结构判断,这个溴正离子的来源只能有一个, 即N-溴代苯甲酰胺的异裂, 其结果必然产生一个溴正离子和一个氮负离子(酰胺基负离子)。对于底物3d, 与双键相连苯环的3, 5-位存在两个强的给电子基团时(CH3O), 会使该苯环的4位电子密度显著增加, 当反应体系中存在亲电试剂(溴正离子)时, 苯环上的4位氢会被溴正离子所取代, 这可能就是5d形成的原因。
 Scheme 4. The formation of the aromatic substituted product 5d
Scheme 4. The formation of the aromatic substituted product 5d
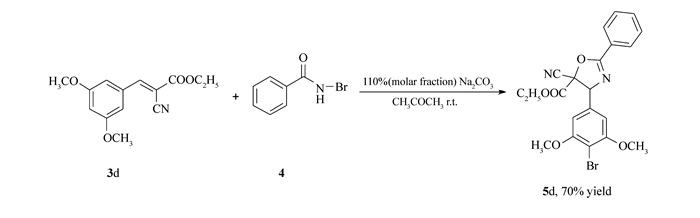
为了进一步探索底物的适应范围,本研究选用α-乙氧甲酰基肉桂酸乙酯6作为反应底物,在相同的反应条件下考察了其与N-溴代苯甲酰胺4的反应情况。实验证明,底物6也能与反应试剂4反应得到相应的2-噁唑啉衍生物7(淡黄色油状),HRMS(C21H22NO5计算值),m/z: 368.1506(368.1498)[M+H]+。但收率有所降低,分离收率37%,见Scheme 5。这一结果说明底物结构中CN的存在并不是该反应的必要条件。
 Scheme 5. Formation of product 7
Scheme 5. Formation of product 7
2.5 反应试剂对反应的影响
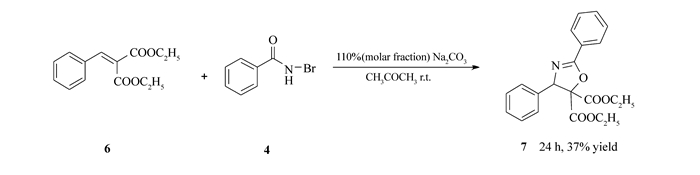
为了进一步探索该反应的普适性,继续以3a(或6)为底物,以N-溴代对硝基苯甲酰胺8和N-溴代乙酰胺9代替反应试剂4,在相同反应条件下考察其反应情况。结果表明,N-溴代对硝基苯甲酰胺8和N-溴代乙酰胺9也可用作该反应的试剂,得到相应的2-噁唑啉衍生物10、11和12,虽然产物10和12的收率不高,见Scheme 6。
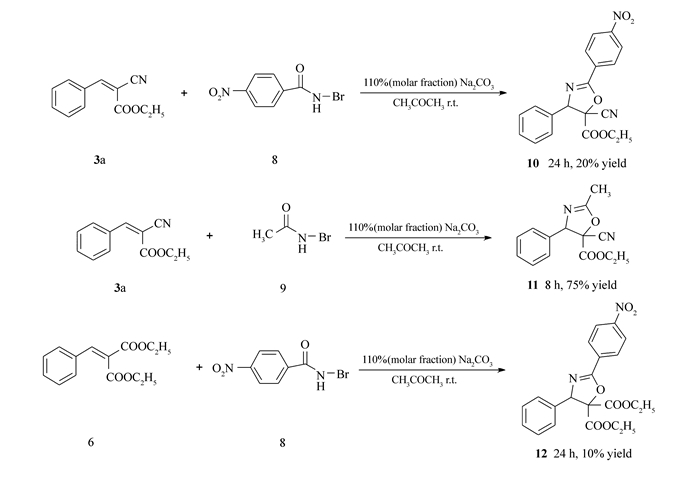 Scheme 6. Different substrates reacted with different reaction reagents
Scheme 6. Different substrates reacted with different reaction reagents
2.6 可能的反应机理
在用TLC跟踪反应进程中发现,在所有的反应中,一开始很快就有一个中间体斑点产生, 随着反应的进行,这个斑点逐渐消失,而另一个新的斑点逐渐生成。反应完成后, 第一个斑点完全消失,而第二个斑点完全形成,经过结构确认第二个斑点是我们所要的最终产物2-噁唑啉衍生物。这一现象使我们意识到, 最终产物2-噁唑啉衍生物并不是一步形成的, 很可能通过一个中间体转化而来。为了证实我们的假设, 我们以3b为底物, 以N-溴代苯甲酰胺4作为反应试剂, 在相同条件下重复实验。约10 min时第一个斑点明显形成, 此时终止实验,分离出第一个斑点,得到中间体13。结构确定该中间体为一个氨溴加成产物, 见图 1。
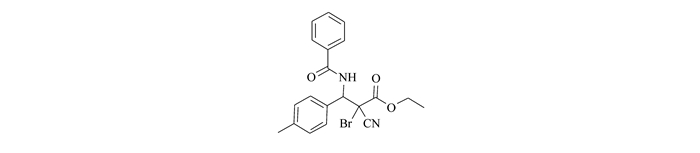 图 1 中间体(13)的结构 Figure 1. Structure of the intermediate(13)
图 1 中间体(13)的结构 Figure 1. Structure of the intermediate(13)
根据产物5d结构中苯环4-位溴的可能来源分析以及对中间体结构的确认, 本研究提出该反应的可能机理如下(见Scheme 7):首先,N-溴代苯甲酰胺4在碱的作用下异裂为溴正离子和苯甲酰氨基负离子(M, Eq.1)。所解离出的溴正离子与α-氰基肉桂酸乙酯3中的双键发生亲电加成反应形成溴鎓离子(A, Eq.2),接下来苯甲酰氨基负离子(M)作为亲核试剂进攻这个溴鎓离子(A),得到一个加成产物(B, Eq. 3)。其所以酰氨基负离子(M)进攻鎓离子(A)中的苄基碳,是因为苯环的存在导致苄基碳所带正电荷较多之故。这个中间体(B, 氨溴加成产物)在碱的作用下脱去酰氨基上一个氢原子,形成一个新的氮负离子,再经过互变异构得到氧负离子(C),接下来氧负离子(C)进攻容易离去的溴发生分子内的亲核取代反应形成五元环2-噁唑啉衍生物5(Eq.4)。
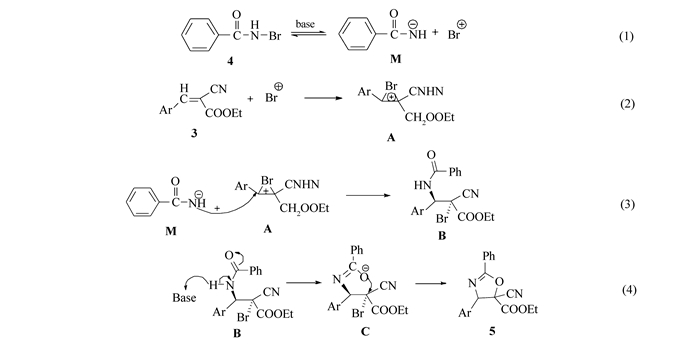 Scheme 7. Possible mechanism for the formation of 2-oxazoline derivatives
Scheme 7. Possible mechanism for the formation of 2-oxazoline derivatives
3 结论
本文以α-氰基肉桂酸乙酯衍生物为底物、以N-溴代苯甲酰胺为反应试剂, 在无水碳酸钠促进下, 以丙酮为溶剂, 室温下建立了合成2-噁唑啉衍生物的新体系。实验结果证明, 在优化条件下, N-溴代苯甲酰胺与不同结构的α-氰基肉桂酸乙酯衍生物可高收率地形成相应2-噁唑啉衍生物, 说明该反应对α-氰基肉桂酸乙酯具有广泛的适应性。实验还证明,N-溴代对硝基苯甲酰胺和N-溴代乙酰胺也能与α-氰基肉桂酸乙酯反应形成相应的2-噁唑啉衍生物,不仅如此,α-乙氧甲酰基肉桂酸乙酯也能与N-溴代苯甲酰胺、N-溴代对硝基苯甲酰胺反应,得到相应的2-噁唑啉衍生物,这表明该反应对底物和反应试剂均具有广泛的适应性。根据实验结果, 本研究提出了该反应的可能机理, 即亲电加加成/分子内亲核取代机理。该机理支持了形成2-噁唑啉衍生物时的区域选择性。
化学慧定制合成事业部摘录




