

亲电芳香族取代是有机反应,其中与芳族体系(通常为氢)连接的原子被亲电试剂取代。一些最重要的亲电芳香取代是芳族硝化,芳族卤化,芳族磺化和酰化和烷基化Friedel-Crafts反应。
说明性反应
该反应最广泛实践的例子是苯的乙基化。

1999年生产了大约24,700,000吨。[2](脱氢和聚合后,生产出商品塑料聚苯乙烯。)在这个过程中,固体酸被用作催化剂以产生初始碳阳离子。苯的许多其他亲电反应被进行,尽管规模小得多,它们是关键中间体的有价值途径。苯的硝化是通过硝鎓离子作为亲电子试剂的作用实现的。所述磺化用发烟硫酸使苯磺酸。与溴,氯的芳香卤化或碘或相应的芳基卤化物。该反应通常由相应的三卤化铁或三卤化铝催化。

该Friedel-Crafts反应可以作为进行酰化或作为烷基化。通常使用三氯化铝,但几乎可以使用任何强的路易斯酸。对于酰化反应,需要化学计算量的三氯化铝。
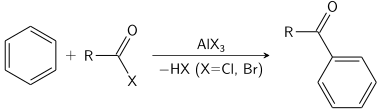

反应机制
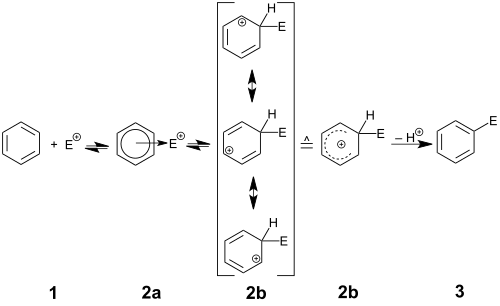
反应机理,表示为S E Ar,以芳环攻击亲电试剂A +开始。该步骤导致形成带正电和离域的环己二烯基阳离子,也称为鎓离子,Wheland中间体或σ络合物。已经表征了该碳阳离子的许多实例,但是在正常操作条件下,这些高酸性物质将附着于sp 3碳的质子供给溶剂(或任何其他弱碱)以重新建立芳香性。最终结果是在芳环中用A取代H. 偶尔,H +旁边的其他电子氧化物将离去重新建立芳香; 这些物质包括甲硅烷基(SiR 3),羧基(CO 2 H),碘基和叔烷基如叔丁基。这些类型的取代基,以留下的容量有时利用合成,特别是通过另一种官能团(取代甲硅烷基的情况下本位取代)。然而,像I +这样的群体的丧失往往是不希望的副反应。
取代基的影响
两个区域选择性 -the不同的可能的芳烃取代模式 -和速度的电芳族取代的由已经连接到苯环上的取代基的影响。在区域选择性方面,一些群体促进了邻位或对位的取代,而其他群体则倾向于在元位置取代。这些组分别称为正交导向或元导向。此外,一些组将增加反应速率(激活),而其他组将降低速率(停用)。虽然区域选择性的模式可以解释共振结构,对动力学的影响可以通过共振结构和诱导效应来解释。
反应速度
关于亲电取代,取代基通常可分为两类:对芳环的活化和失活。 活化取代基或活化基团通过感应效应或共振效应将电子给予环系统来稳定在取代期间形成的阳离子中间体。活化芳环的实例是甲苯,苯胺和苯酚。
通过取代基递送到环中的额外电子密度不是均匀地分布在整个环上,而是集中在原子2,4和6上,因此活化取代基也是邻位/对位指导(见下文)。
另一方面,使取代基失活使中间阳离子不稳定,从而通过诱导或共振效应降低反应速率。它们是通过从芳环中提取电子密度来实现的。芳族体系的失活意味着通常需要更苛刻的条件来促使反应完成。这方面的一个例子是在三硝基甲苯的生产过程中甲苯的硝化(TNT)。在活化的甲苯环上进行的第一次硝化可以在室温下进行,并且使用稀酸,第二次硝化,在去活的硝基甲苯环上,已经需要长时间加热和更浓的酸,第三次,非常强烈地失活二硝基甲苯,必须在沸腾浓硫酸中进行。通过共振吸电的基团降低电子密度,特别是在位置2,4和6处,使位置3和5作为具有相对较高反应性的那些,因此这些类型的基团是间位导向剂(参见下文)。卤素是带负电的,因此它们通过诱导失活,但它们只有一对,因此它们是共振供体,因此是邻位/对位指导。
具有未共享电子对的基团,例如苯胺的氨基,通过共振强烈活化和邻位/对位取向。这些激活基团将那些未共享的电子捐赠给pi系统,在邻位和对位产生负电荷。因此,这些位置对于缺电子的亲电试剂最具反应性。最高的电子密度位于邻位和对位,[ 需要澄清 ]尽管这种增加的反应性可能被空间位阻所抵消取代基和亲电试剂之间。因此,亲电子芳族取代的最终结果可能难以预测,并且通常仅通过进行反应并确定邻位与对位取代的比率来确定。

除了原环的亲核性质增加外,当亲电试剂攻击苯胺的邻位和对位时,氮原子可以向pi系统提供电子密度(形成亚胺离子),产生四种共振结构(相对于三个在基本反应中)。这显着提高了阳离子中间体的稳定性。

当亲电试剂攻击元位时,氮原子不能向pi系统提供电子密度,仅给出三个共振贡献者。这种推理与间位取代产物的低产率一致。

其他取代基,例如烷基和芳基 取代基,也可以向π系统提供电子密度; 然而,由于它们缺少可用的非共享电子对,因此它们的能力相当有限。因此,它们仅微弱地激活环并且不会强烈地不利于元位置。
定向邻位金属化是一种特殊类型的具有特殊邻位导向的EAS 。
具有比碳更具电负性的原子的非卤素基团,例如羧酸基团(-CO 2 H),从π系统中取出大量电子密度。这些群体强烈停用群组。另外,由于取代的碳已经贫电子,具有谐振贡献者的任何结构,其中有对碳轴承他吸电子基团具有正电荷(即,邻位或对位攻击)比其它的更不稳定。因此,这些吸电子基团是元导向的,因为这是不具有那么多不稳定性的位置。
反应也慢得多(相对于苯的相对反应速率为6×10 -8),因为环的亲核性较低。
对吡啶的反应
与苯相比,由于氮原子的电负性较高,吡啶上的亲电取代速率要慢得多。另外,吡啶中的氮容易通过质子化(来自硝化或磺化)或用于催化反应的路易斯酸(例如AlCl 3)获得正电荷。这通过在碳和氮上具有相邻的形式电荷或在局部原子上具有2个形式电荷而使反应更慢。直接在吡啶中进行亲电取代几乎是不可能的。
为了进行反应,它们可以通过两种可能的反应制备,这两种反应都是间接的。
在吡啶上进行取代的一种可能方法是亲核芳族取代。即使没有催化剂,具有电负性的氮原子本身也可以保持负电荷。另一种方法是在亲电取代之前进行氧化。这使得由氧负氧原子引起的吡啶N-氧化物使反应比吡啶,甚至苯更快。然后氧化物可以还原成取代的吡啶。
Ipso
进入基团与已经带有取代基(除氢之外)的芳族化合物中的位置的连接。进入的基团可以取代该取代基,但也可以在随后的步骤中将其排出或迁移到不同的位置。不使用术语’ ipso -substitution’,因为它与替换同义。一个典型的例子是水杨酸与硝酸和硫酸的混合物反应形成苦味酸。2位的硝化涉及作为离去基团的CO 2的损失。其中磺酰基被质子取代的脱磺酸作用是常见的例子。也可以看看林重排。在被硅取代的芳族化合物中,硅通过取代反应而发生反应。
五元杂环
与苯相比,呋喃,噻吩和吡咯更易受亲电子攻击。这些化合物都含有具有未共用电子对(氧,硫或氮)的原子作为芳环的成员,其基本上稳定了阳离子中间体。吡咯的亲电取代的实例是Pictet-Spengler反应和Bischler-Napieralski反应。
不对称亲电芳香取代
通过转换为手性路易斯酸催化剂,特别是在Friedel-Crafts型反应中,用前手性碳亲电试剂进行的亲电芳族取代已经适用于不对称合成。一个早期的例子是关于加入三氯乙醛至酚催化氯化铝与改性( – ) -薄荷醇。[4]甲乙醛酸化合物已被添加到N,N-二甲基苯胺与手性的二恶唑啉配位体 – 铜(II),三氟甲磺酸催化剂体系也以 Friedel-Crafts羟烷基化:[5]

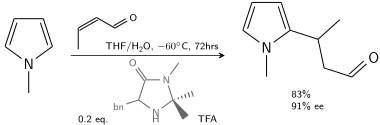
在另一种烷基化中,N-甲基吡咯与用手性咪唑烷酮改性的三氟乙酸催化的巴豆醛反应:[6]
吲哚与手性BINOL衍生的磷酸催化的烯酰胺反应:[7]
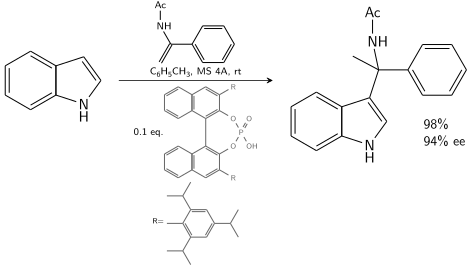
在10-20%手性催化剂存在下,可实现80-90%ee。
其他反应
- 在亲电芳族取代模式之后的其他反应是一组芳族甲酰化反应,包括Vilsmeier-Haack反应,Gattermann Koch反应和Reimer-Tiemann反应。
- 其他亲电子是芳族重氮盐在重氮偶合,二氧化碳在科尔比-施密特反应和活化的羰基在pechmann缩合反应,hydroxycarbenium离子在布兰克氯甲基化反应通过中间(羟甲基)芳烃(苄醇),chloryl阳离子(CLO 3 +)用于亲电子的perchlorylation。
- 在多步骤Lehmstedt-Tanasescu反应中,其中一个亲电子试剂是N-亚硝基中间体。
- 在Tscherniac-Einhorn反应(以Joseph Tscherniac和Alfred Einhorn命名)中,亲电子试剂是酰胺的N-甲醇衍生物




