

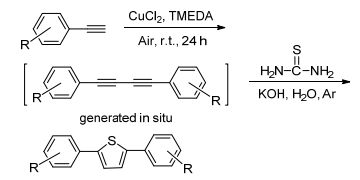
 图式1 2, 5-二芳基噻吩的合成设计 图式1. Design for syntheses of 2, 5-diarylthiophene
图式1 2, 5-二芳基噻吩的合成设计 图式1. Design for syntheses of 2, 5-diarylthiophene 噻吩是一个基本五元芳香杂环, 含噻吩结构的天然产物大多具有良好的生物活性, 而且噻吩衍生物的聚合物在现代光电子、微电子领域及现代医学等领域中也具有诱人的应用前景, 因而新噻吩衍生物和新合成方法的研究一直受到关注[1].噻吩及其衍生物的经典制备方法是Paal-Knorr反应, Paal-Knorr反应是利用1, 4-二羰基化合物与含硫化合物缩合生成噻吩及其衍生物[2].此外, 化学工作者也在探索以其它化合物为原料的噻吩及其衍生物的合成方法[3], 经探索发现1, 3-丁二炔衍生物作为一种重要的合成中间体, 可以用于各种环状化合物的合成[4], 而且使用1, 3-丁二炔及其衍生物作为原料时, 反应具有原子经济性、原料简单易得等特点.因而, 近来有一批文献报道了一些以1, 3-丁二炔衍生物为原料的合成噻吩及其衍生物的新方法[5].例如Hua等[5a]报道了钯催化或者直接采用氢氧化钾促进炔烃与Na2S反应, 高产率地合成对称的2, 5-二取代噻吩的方法. Zhang等[5b]报道了碱促进的中间体S3自由基参与的1, 3-丁二炔及其衍生物环化生成2, 5-二取代噻吩的合成方法, 该方法操作简单, 产物易分离. Zhao等[5c]报道了NaHS可以在无金属催化剂存在的条件下与1, 3-丁二炔反应高产率地生成2, 5-二取代噻吩.尽管利用上述方法已经可以有效合成噻吩类化合物, 但鉴于噻吩化合物在实际应用中的重要性, 我们认为发展从简单易得原料出发, 更加简单高效、绿色合成取代噻吩杂环的合成方法仍然很有必要.因此, 本课题组将报道一个绿色的, 高效地合成2, 5-二取代噻吩的新方法(Scheme 1), 并将对合成出噻吩衍生物进行初步的光学性质研究.
图式1 2, 5-二芳基噻吩的合成设计 图式1. Design for syntheses of 2, 5-diarylthiophene
1 结果与讨论
1.1 最佳反应条件的建立
我们采用苯乙炔作为反应原料经过偶联、环化等反应串联合成了2, 5-二苯基噻吩.实验过程中首先以空气为氧化剂, 以铜化合物为催化剂, 在室温无溶剂的条件下, 苯乙炔经过24 h氧化偶联得到1, 3-二炔类化合物; 然后不经分离, 在反应混合物中加入水为溶剂, 以TEBA为相转移催化剂, 以硫代有机物、NaHS为硫源, 在Ar保护碱性条件下, 于120 ℃经过一定时间的反应, 环化生成2, 5-二取代噻吩化合物.探索了合成2, 5-二取代噻吩的反应条件.实验结果见表 1.
 |
|||||
| Entry | Catalyst | Ligand | Base | Reaction time/h | Yieldb/% |
| 1 | CuCl2 | TMEDA | KOH | 24, 24 | 71.0 |
| 2 | CuI | TMEDA | KOH | 24, 24 | 46.6 |
| 3 | CuCl | TMEDA | KOH | 24, 24 | 29.0 |
| 4 | Cu2O | TMEDA | KOH | 24, 24 | 22.4 |
| 5 | Cu(acac)2 | TMEDA | KOH | 24, 24 | 13.8 |
| 6 | CuCl2 | TEA | KOH | 24, 24 | 53.9 |
| 7 | CuCl2 | Oxine | KOH | 24, 24 | 48.4 |
| 8 | CuCl2 | 1, 10-Phenanthroline | KOH | 24, 24 | 46.2 |
| 9 | CuCl2 | Methenamine | KOH | 24, 24 | 45.4 |
| 10 | CuCl2 | TMEDA | NaOH | 24, 24 | 49.9 |
| 11 | CuCl2 | TMEDA | NaOAc | 24, 24 | 34.0 |
| 12 | CuCl2 | TMEDA | K2CO3 | 24, 24 | 47.7 |
| 13c | CuCl2 | TMEDA | KOH | 24, 24 | 76.1 |
| 14d | CuCl2 | TMEDA | KOH | 24, 24 | 45.0 |
| 15c | CuCl2 | TMEDA | KOH | 24, 36 | 89.3 |
| a General reaction conditions: phenylacetylene (2 mmol), Cu compounds (1 mol%), ligand (5 mol%), room temperature; base (2.0 mmol, cyclic reaction), H2O (1.0 mL, cyclic reaction), TAA (1.5 mmol, cyclic reaction), under Ar (cyclic reaction), TEBA (5 mol%, cyclic reaction), 120 ℃ (cyclic reaction). b Isolated yield. c 1.5 mmol of thiourea was used in place of TAA.d 1.5 mmol of NaHS was used in place of TAA. | |||||
从表 1中的数据可以看出, 二氯化铜的催化效果优于其它铜盐(表 1, Entries 1~5).随后, 我们以二氯化铜为催化剂, 考察了各种配体对反应的影响.实验结果显示以TMEDA为辅助配体时, 反应收率最高, 达71.0% (表 1, Entries 1, 6~9).接着, 考察了各种碱对反应的影响.在被考察的碱中, KOH得到了最高的收率, 而其它碱虽有一定效果, 但与KOH相比, 结果都不太理想(表 1, Entries 1, 10~12), 因此, 在后续反应中, 我们均以KOH作为碱来考察其它因素对反应的影响.考察不同硫源对反应的影响结果显示, 相对于硫代乙酰胺, 硫尿作为硫源时, 反应收率更高, 达到76.1%(表 1, Entry 13);而使用NaHS作为硫源时, 反应收率只有45% (表 1, Entry 14).最后考察了时间对该反应的影响, 尝试延长环化反应时间到36 h, 产率升高到89.3%(表 1, Entry 15).
1.2 2, 5-二苯基噻吩衍生物的合成
在得到了合成2, 5-二苯基噻吩的最佳反应条件之后, 我们进一步探索了不同芳基端炔在该反应条件下的反应活性, 实验结果见表 2.
 |
从表 2中的数据可以看出, 在该反应条件下, 以各种芳基端炔为原料的反应均可以高效地合成2, 5-二取代噻吩, 实验结果也显示, 当苯环对位上有强供电子基团时, 反应的收率有所降低(表 2, Entries 3, 5);当苯环的邻、对位上有弱供电子基时, 反应的收率变化不大(表 2, Entries 1, 2, 4, 7);当苯环被吡啶环取代时, 反应的收率变化也不大(表 2, Entry 8).实验结果还显示苯环间位上的氨基对反应也没有明显的影响(表 2, Entry 6).
1.3 可能的反应机理
根据文献[5b, 6], 我们认为该合成过程是由两步串联而成, 第一步是端炔在铜催化下氧化偶联生成1, 3-丁二炔, 第二步是硫尿在碱作用下形成S-离子, S-离子通过两次加成-环化生成2, 5-二取代噻吩.具体的反应机理见Scheme 2.
 图式2 可能的反应机理 图式2. Possible reaction mechanism
图式2 可能的反应机理 图式2. Possible reaction mechanism
1.4 2, 5-二苯基噻吩类化合物的光学性质
1.4.1 2, 5-二苯基噻吩类化合物的紫外光谱
从上述合成出的8种2, 5-二取代噻吩化合物中选择化合物2a, 2e~2i为代表, 研究了2, 5-二取代噻吩化合物的紫外光谱. 图 1为所选择的化合物在CH3OH (1×10-5 mol/L)中的紫外-可见吸收光谱(UV-Vis).
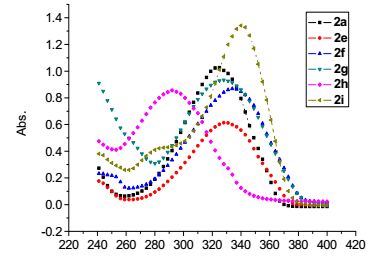 图 1 化合物2a和2e~2i在甲醇中紫外吸收谱图(1×10-5 mol/L) Figure 1. UV-Vis spectra of compounds 2a and 2e~2i in methanol (1×10-5 mol/L)
图 1 化合物2a和2e~2i在甲醇中紫外吸收谱图(1×10-5 mol/L) Figure 1. UV-Vis spectra of compounds 2a and 2e~2i in methanol (1×10-5 mol/L)
如图 1所示, 所选择的化合物2a, 2e, 2f, 2g, 2h和2i的紫外最大吸收波长位于292~341 nm之间, 这一范围的吸收是由共轭体系中噻吩环和芳环共轭的π-π*跃迁引起的, 由于芳环的不同, 或由于苯环上取代基的不同, 导致了所选择的化合物2a, 2e~2i的紫外最大吸收波长在292~341 nm之间波动. 2a的最大紫外吸收波长为324 nm, 2e相对2a来说, 苯基的4-位上有甲基取代基, 甲基与苯环上的π66键存在着超共轭效应, 2e的最大紫外吸收波长小幅向长波方向移动, 为329 nm. 2f相对于2a来说, 苯基的4-位上有甲氧基取代基, 甲氧基中的氧上的p电子对与苯环上的π66键存在着共轭效应, 2f的最大紫外吸收波长向长波方向移动, 为334 nm. 2g相对于2a来说, 苯基的3-位上有氨基取代基, 氨基中的氮上的p电子对与苯环上的π66键存在着共轭效应, 2g的最大紫外吸收波长向长波方向移动, 为329 nm. 2h相对2a来说, 苯基的2-位上有甲基取代基, 由于甲基的体积位阻较大, 导致了苯环与噻吩环不能很好地共平面, 破坏了苯环与噻吩环的共轭效应, 2e的最大紫外吸收波长向短波方向移动, 为292 nm. 2i相对于2a来说, 苯环被置换成了吡啶环, 2i的最大紫外吸收波长为341 nm.
1.4.2 2, 5-二苯基噻吩类化合物的荧光光谱
用甲醇、二氯甲烷分别作溶剂将2a, 2e~2i配成5×10-6 mol/L的浓度, 测得紫外吸收光谱, 分别得到相应的最大吸收波长和它对应的吸光度值A, 再用最大吸收波长作为激发波长, 测定它们的发射光谱λEM1, 再以λEM1最大值测定激发光谱λEX, 最后再以λEX最大值测定目标产物的荧光发射光谱λEM2, 测定结果如图 2和图 3所示. 图 2为上述化合物在甲醇中的荧光发射光谱, 从图 2中可以看出, 上述化合物的最大发射波长在386~454.5 nm之间, 化合物2e, 2f, 2g和2i的最大荧光发射波长相比2a的最大发射波长发生了红移, 尤其是2g的红移(67 nm)最为明显, 这可能是取代基使共轭体系增大的缘故; 2h的最大荧光发射波长相比2a的最大发射波长, 发生了蓝移, 这可能是由于甲基在苯环的2位时, 位阻破坏了苯环与噻吩环的有效共轭. 图 3为上述化合物在二氯甲烷中的荧光发射光谱, 从图 3中可以看出, 上述化合物的最大发射波长在390~412 nm之间, 化合物2e, 2f, 2g和2i的最大荧光发射波长相比2a的最大发射波长发生了红移, 这可能是取代基使共轭体系增大的缘故; 2h的最大荧光发射波长相比2a的最大发射波长发生了蓝移, 这可能是由于2位上的甲基的位阻使苯环与噻吩环不能有效共轭.从实验结果可以发现, 上述化合物在甲醇中荧光强度要强一些, 当苯环3位上取代基是氨基时, 化合物的最大荧光发射波长在甲醇中的红移明显, 在二氯甲烷中只有轻微的红移.
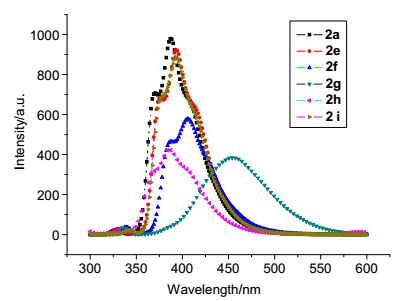 图 2 化合物2a和2e~2i在甲醇中的荧光光谱(5×10-6 mol/L) Figure 2. Fluorescence spectra of compounds 2a and 2e~2i in methanol (5×10-6 mol/L)
图 2 化合物2a和2e~2i在甲醇中的荧光光谱(5×10-6 mol/L) Figure 2. Fluorescence spectra of compounds 2a and 2e~2i in methanol (5×10-6 mol/L)
 图 3 化合物2a和2e~2i在二氯甲烷中的荧光光谱(5×10-6 mol/L) Figure 3. Fluorescence spectra of compounds 2a and 2e~2i in dichloromethane (5×10-6 mol/L)
图 3 化合物2a和2e~2i在二氯甲烷中的荧光光谱(5×10-6 mol/L) Figure 3. Fluorescence spectra of compounds 2a and 2e~2i in dichloromethane (5×10-6 mol/L)
2 结论
本文以芳基乙炔与硫脲为起始原料, 通过一锅法, 经过氧化偶联反应和成环反应两步串联, 合成了2, 5-二苯基噻吩及其衍生物.在此基础上, 进一步探索了所合成化合物的UV和荧光性质, 实验结果表明所合成化合物在甲醇中的紫外最大吸收波长在292~341 nm之间, 荧光光谱表明该类化合物具有良好的荧光性, 其在甲醇中最大发射波长在386~454.5 nm之间, 在二氯甲烷中测定的最大发射波长在390~412 nm之间.
3 实验部分
3.1 仪器与试剂
高分辨质谱测试: AB SCIEX Triple 5600+型液质连用仪(美国AB SCIEX公司). 1H NMR和13H NMR谱: Mercury Plus 400型核磁共振仪(美国Varian公司), CDCl3为溶剂, TMS为内标.紫外吸收光谱: Lambda 950紫外可见分光光度计上测定, 测试溶液样品使用的样品池是通光长度为1.0 cm的两面通光的石英池.荧光光谱测试:带恒温系统的LS-50B型荧光光度计(美国Perkin-Elmer公司)测定, 测试溶液样品使用的样品池为1.0 cm×1.0 cm四面通光的石英比色皿.
所用的化学原料试剂全部从Alfa、Aldrich、百灵威等公司购买, 没有进一步的纯化; 常用溶剂购自国药集团; 薄层层析硅胶由硅胶GF254 (青岛海洋化工厂)和羧甲基纤维素(CMC)自制; 柱层析用硅胶为化学纯试剂(青岛海洋化工厂).
3.2 实验方法
3.2.1 2, 5-二取代噻吩的合成
往反应管中依次加入搅拌磁子、末端炔(2 mmol)、CuCl2 (5 mol%)和四甲基乙二胺(TMEDA) (5 mol%), 反应在室温搅拌反应24 h; 然后加入2 mL纯水、硫尿(1.5 mmol), TEBA (5 mmol%)和KOH (2 mmol), 升温至120 ℃下搅拌反应36 h至薄层色谱监测原料完全转化, 直接中性氧化铝拌样经柱层析纯化, 使用V(石油醚):V(二氯甲烷)=15:1作为淋洗剂, 得到产物2.
2, 5-二苯基噻吩(2a)[7]: 211 mg, 白色固体, m.p. 150~152 ℃ (lit.[7] 153 ℃); 1H NMR (CDCl3, 400 MHz) δ: 7.26~7.30 (m, 4H, ArH), 7.39 (t, J=8.0Hz, 4H, PhH), 7.64 (d, J=7.6 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 143.6, 134.3, 128.9, 127.5, 125.7, 124.0; HRMS (ESI) calcd for C16H13S [M+H]+ 237.0738, found 237.0733.
2, 5-二(4′-乙基苯基)噻吩(2b): 211 mg, 淡黄色固体, m.p. 170~172 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.25 (t, J=7.6 Hz, 6H, CH3), 2.66 (q, J=7.6 Hz, 4H, CH2), 7.20 (s, 2H, ArH), 7.23 (t, J=7.6 Hz, 4H, PhH), 7.54 (d, J=8.0 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 143.7, 143.3, 131.9, 128.4, 125.6, 123.5, 28.6, 15.6; HRMS (ESI) calcd for C20H21S [M+H]+ 293.1364, found 293.1358.
2, 5-二(4′-丁基苯基)噻吩(2c): 245 mg, 淡黄色固体, m.p.130~131℃ (lit.[5c] 132~134 ℃); 1H NMR (CDCl3, 400 MHz) δ: 0.93 (t, J=7.6Hz, 6H, CH3), 1.34~1.38 (m, 4H, CH2), 1.59~1.63 (m, 4H, CH2), 2.63 (q, J=7.6 Hz, 4H, CH2), 7.19 (d, J=8.0 Hz, 4H, PhH), 7.23 (s, 2H, ArH), 7.53 (d, J=8.4 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 13.98, 22.37, 33.56, 35.37, 123.43, 125.51, 128.95, 131.87, 142.37, 143.29; HRMS (ESI) calcd for C24H29S[M+H]+ 349.1990, found 349.2011.
2, 5-二(4′-乙氧基苯基)噻吩(2d): 176 mg, 白色固体, m.p. 216~218 ℃; 1H NMR (CDCl3, 400 MHz) δ: 1.43 (t, J=7.2 Hz, 6H, CH3), 4.06 (q, J=7.2 Hz, 4H, CH2), 6.91 (d, J=8.8 Hz, 4H, PhH), 7.14 (s, 2H, ArH), 7.53 (d, J=8.8 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 14.9, 63.6, 114.9, 122.8, 126.8, 127.2, 142.6, 158.6; HRMS (ESI) calcd for C20H21O2S[M+H]+ 325.1262, found 325.1254.
2, 5-二(4′-甲基苯基)噻吩(2e)[4p]: 227 mg, 白色固体, m.p. 157~159 ℃ (lit.[8] 161~164 ℃); 1H NMR(CDCl3, 400 MHz) δ: 2.37(s, 6H, CH3), 7.19 (d, J=7.6 Hz, 4H, PhH), 7.23 (s, 2H, ArH), 7.52 (d, J=8.0 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 21.2, 123.4, 125.5, 129.6, 131.7, 137.3, 143.3; HRMS (ESI) calcd for C18H17S[M+H]+ 265.1051, found 265.1049.
2, 5-二(4′-甲氧基苯基)噻吩(2f)[5c]: 164 mg, 白色固体, m.p. 192~194 ℃ (lit.[5c] 216~217 ℃); 1H NMR (CDCl3, 400 MHz) δ: 3.84 (s, 6H, CH3), 6.92 (d, J=8.8 Hz, 4H, PhH), 7.25 (s, 2H, ArH), 7.54 (d, J=8.4 Hz, 4H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 55.39, 114.31, 122.90, 126.84, 134.06, 142.60, 159.12; HRMS (ESI) calcd for C18H17O2S[M+H]+ 297.0949, found 297.0947.
2, 5-二(3′-氨基苯基)噻吩(2g)[7]: 198 mg, 白色固体, m.p. 202~204 ℃ (lit.[7] 202 ℃); 1H NMR (CDCl3, 400 MHz) δ: 3.72 (s, 4H, NH2), 6.60~6.62 (m, 2H, PhH), 6.94 (m, 2H, PhH), 7.02~7.05 (m, 2H, PhH), 7.14~7.18 (m, 2H, ArH), 7.22(s, 2H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 112.2, 114.4, 116.2, 123.8, 129.6, 135.7, 143.6, 146.6; HRMS (ESI) calcd for C16H15N2S[M+H]+ 267.0956, found 267.0953.
2, 5-二(2′-甲基苯基)噻吩(2h)[7]: 190 mg, 无色液体. 1H NMR (CDCl3, 400 MHz) δ: 2.50 (s, 6H, CH3), 7.06 (s, 2H, ArH), 7.24~7.28 (m, 6H, PhH), 7.47 (m, 2H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 21.3, 126.0, 126.5, 127.8, 130.3, 130.7, 134.1, 136.0, 142.9; HRMS (ESI) calcd for C18H17S [M+H]+ 265.1051, found 265.1047.
2, 5-二(2′-吡啶基)噻吩(2i)[5c]: 167 mg, 淡黄色固体, m.p. 154~157 ℃ (lit.[5c] 157~159 ℃; 1H NMR (CDCl3, 400 MHz) δ: 7.16~7.20 (m, 2H, ArH), 7.65~7.75 (m, 6H, PhH), 8.60 (d, J=4.8 Hz, 2H, PhH); 13C NMR (CDCl3, 100 MHz) δ: 112.2, 114.4, 116.2, 123.8, 129.6, 135.4, 146.6. HRMS (ESI) calcd for C14H11N2S [M+H]+ 239.0643, found 239.0641.
化学慧定制合成事业部摘录




